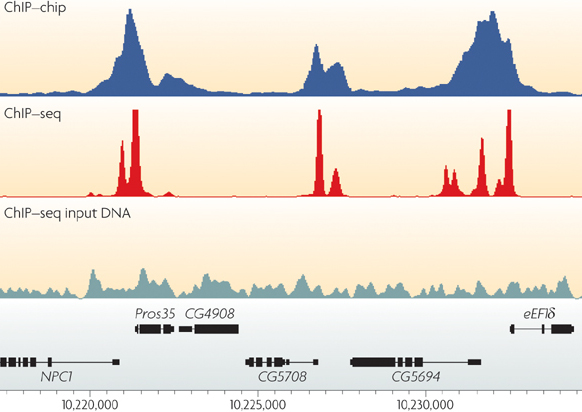
Adapted from: ChIP–seq: advantages and challenges of a maturing technology
Peter J. Park
Nature Reviews Genetics 10, 669-680 (October 2009)
One of the most common sequencing applications searched for and ordered on Genohub is ChIP-Seq. A frequent question we’re asked is how much sequencing do I need for my ChIP-Seq experiment?
ChIP-Seq is the most widely used technique for measuring protein – DNA interactions on a genome wide scale. Before starting a ChIP-Seq experiment it’s important to have some information about specificity. Assume that you’ve tested your protein’s (nucleosomes, histones, chaperone) specificity and it enriches 10x over background. If you’re fragmenting a human genome and there are ~3,000 places in the genome that your protein binds, you’ll need approximately 1 sample/fragment:
Your background (human) is : 3 Gb / 300 bp, or 1×107 fragments.
Signal enrichment is 10x x 3000 locations = 3×104
So you need 1×107 + 3×104 ~= 1×107 sample hits for a 10x signal, with 10 fold enrichment.
Here are some services on Genohub that would meet these ChIP-Seq metrics: https://genohub.com/shop-by-next-gen-sequencing-technology/#query=cac50ae846ca1cf773a39716b66f7142.
ChIP Signal Strength
The relationship between ChIP signal strength and regulatory activity is an area of active research. Some very active transcriptional enhancers often display moderate ChIP signal. As a result it can be difficult to set a threshold for Chip Signal strength that will be inclusive of all functional sites. A rough guide is ~ 20M unique mapped reads / mammalian sample.
ChIP-Seq Control
Designing a good control is essential for every ChIP-Seq experiment. A separate control should be run for every sample, cell type, condition or treatment. For a useful control, perform ChIP with an antibody that reacts with an unrelated antigen. Make sure you’re able to make a library that’s as complex as your experimental samples. We typically recommend that users dedicate at least the same if not more reads to their control versus actual samples.
Making highly ‘complex’ libraries is important for ChIP-Seq. We’ve outlined several library prep kit options here: https://genohub.com/chip-seq-library-preparation/.
Finally, if you’re new to ChIP-Seq and need more project advice, contact us for complimentary project consultation.

